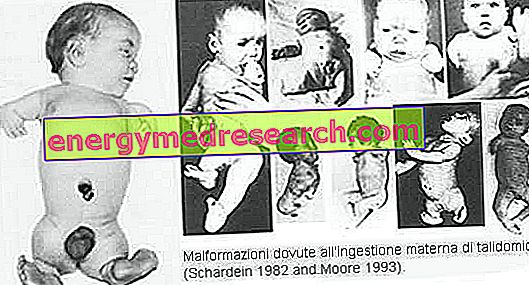
관련 기사 : 불완전한 골 형성 정의 불완전한 골 형성은 뼈의 취약성과 다양한 골격 변형을 특징으로하는 일련의 유전병을 포함합니다. 현재 방사선학적인 특징과 분자 유전 학적 분석을 토대로 15 가지 유형을 구별합니다. 불완전한 골 형성의 주된 형태는 4 가지 (유형 I-IV); 유형 I과 IV는 상 염색체 우성 방식으로 전달되는 반면, 유형 II와 III는 상 염색체 열성 형으로 전달됩니다. 다른 형태는 다소 드물다. 이 경우의 95 %에서 기원은 COL1A1 및 COL1A2 유전자의 돌연변이 (각각 I 형 콜라겐의 알파 -1 및 알파 -2 사슬을 코딩 함)에 기인한다. 가장 흔한 증상 및 징후 * 뼈 통증 합동 통증 성장의 고통 근육통 멍 뼈 골절 수경 태아 hyperkyphosis 전만증 합동과 운동성 청력 손실 대두 증 태아 사망 골감소증 카네이션 처리 된 유방 성장 지연 깔때기 모양의 가슴 추가 징후 불완전한 골 형성은 매우 다양한 임상 사진으로 발생합니다. 일반적으로이 상태는